Mass spectrometry
The precise mass of intact peptides and proteins derived from them can be determined through mass spectrometry. This is a very sensitive method which needs only very small amounts of material. The most generally used mass spectrometric techniques is called MALDI-TOF (matrix-assisted laser desorption ionization- time-of-fiight spectrometry). In this technique peptides are first mixed with an organic acid and then dried onto a metal slide or ceramic. The sample is then blasted with a laser that causes the peptides to be ejected from the slide in the form of an ionized gas in that each molecule carries one or more positive charges. Ionized peptides are then accelerated in an electricfiy and field toward a detector. Overall time it takes for them to reach the detector is determined through their mass and their charge; large peptides move more slowly and more highly charged peptides move more fastly. The precise mass is then determined through analysis of those peptides with a single charge. If a combination of tryptic peptides is used then the resulting masses measured in the MALDI-TOF can be used to find protein sequence databases for matches with theoretical massess calculated for all trypsin-released peptides for all proteins in a sequenced genome. Although, the identity of the original protein and its sequence can readily be determined through a combination of mass protein sequence and spectrometry database seeking.
A differentiation of this technique can be used straightly to determine the sequences of the individual peptides. Subsequently trypsin digestion of the purified protein and determination of their masses through mass spectrometry as above, every peptide is further fragmented at the peptide bonds and the masses of these fragments measured in a coupled second mass spectrometer. This is so-called tandem MS-MS (mass spectrometry). The mass differences among the fragments can be used to construct a partial amino acid sequence in that, in turn, can be used to find protein sequence databases or gives the means for cloning the gene.
Sequencing of proteins through mass spectrometry has many benefits over traditional chemical Edman sequencing:
- Material are needed for much smaller amounts.
- The series of the peptide can be acquired in only a few minutes compared with the hour required for just one cycle of Edman degradation.
- Mass spectrometry can be used to sequence various polypeptides in a mixture, alleviating the need to completely purify the sample prior to analysis.
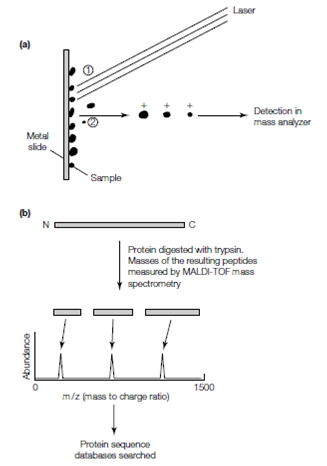
Figure: Mass spectrometry to determine protein sequence. (a) In a MALDI-TOF mass spectrometer, pulses of light from a laser ionize a peptide mixture that is absorbed on a metal slide (1). An electric ?eld accelerates the molecules in the sample toward the detector (2). The time to the detector is inversely proportional to the mass of the protein. (b) Use of mass spectrometry to identify proteins. The protein of interest is digested with trypsin and the resulting peptide fragments are loaded into the mass spectrometer where their masses are measured. Sequence databases are then searched to identify the protein whose calculated tryptic digest pro?le matches the experimentally determined data.
- the Mass spectrometry should be used to determine the sequence of peptides which have blocked N-termini, like as pyroglutamate, a derivative of glutamate in that the side-chain carboxyl group forms an amide bond with its primary amino group (a common eukaryotic post-translational modi?cation which prevents Edman degradation) and to characterize other post- translational modi?cations like as phosphorylation and glycosylation.