Human TSEs
Prior to the mid 1990s there were four forms of human TSE identified; Creutzfeldt-Jakob disease (CJD), the rare, inherited fatal familial insomnia (FFI), Gerstmann-Straussler- Scheinker disease (GSS), and the now extinct Kuru, which was the first human TSE to undergo extensive investigation. The first cases were recorded in the 1950s and occurred in male and female adolescents and adult women members of the Fore people in the highlands of New Guinea and at one point was the leading cause of death of women of the tribes. The Fore people practiced ritual cannibalism as a rite of mourning for their dead, the women and children but not the adult men taking part in the ceremony. The disease demonstrated progressive loss of voluntary neuronal control followed by death within 1 year after the onset of symptoms. As the cannibalistic ritual ceased so did transmission of the disease and although some individuals developed symptoms after an incubation period of up to 30 years (usually it was much shorter), the disease is now eradicated. Kuru demonstrated clearly that a human TSE could be acquired via the oral route.
Patients with CJD present with a progressive subacute or chronic decline in cognitive or motor function associated with recurrent seizures. CJD is rare, with an annual worldwide incidence of approximately one per million populations and can be familial (inherited;
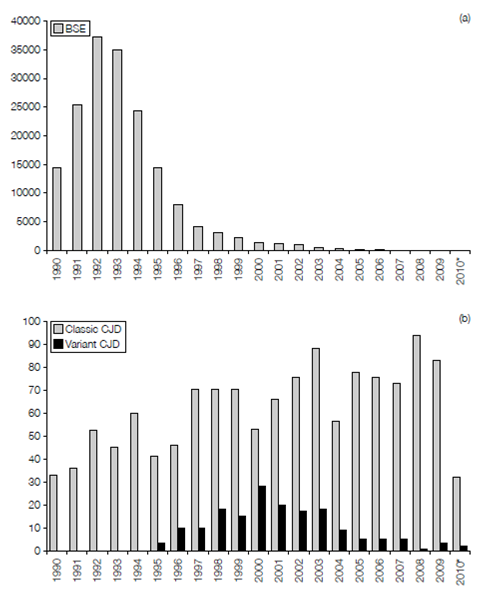
Figure: Reported incidence of (a) BSE and (b) vCJD in the UK between 1990 and September 2010 (i.e. partial data for 2010* only). Data obtained from the World Organization for Animal Health and the National CJD Surveillance Unit, University of Edinburgh. For comparison purposes, classic CJD includes the combined annual figures for familial, iatrogenic, and sporadic CJD shown in chart b.
10% of cases), or acquired sporadically or iatrogenically (cases have been recorded fol- lowing corneal grafts, the administration of pituitary hormones or the use of electrodes contaminated with PrPsc).
In April 1996 a new variant of CJD (vCJD) was described in the UK. The disease has fea- tures that distinguish it from other forms of CJD including an early age of onset (average 27 years as opposed to 65 for CJD), suggesting a shorter incubation period, a prolonged period of illness (average 13 months as opposed to 3 months for CJD), and a psychiatric presentation as opposed to neurological symptoms, with the absence of the typical electroencephalography (EEG) appearances of CJD. In terms of pathology vCJD resembles Kuru, particularly the type of amyloid plaque formation. Evidence has accumulated that the BSE prion has been transferred (most likely via the oral route) to humans and is responsible for vCJD. The source of the infected material is unknown, although BSE- contaminated cattle products are a likely candidate. In 1989 the human consumption of specified bovine offal (brain, spleen, thymus, tonsil, and gut) was prohibited in the UK and in 1996 the ban was extended to sheep offal. Furthermore, the brain and CNS are now removed from all cattle at abattoirs before the distribution of meat.
Up to September 2010, 118 cases of deaths as a result of vCJD have been confirmed, with a further 51 deaths likely to be due to this TSE (post-mortem confirmation pending). There is strong epidemiological evidence that the epidemic has peaked (Figure 2) but it is worth considering that all but one case to date have occurred in individuals homozygous for valine at the position 129 polymorphism described above. Since all forms of the poly- morphism support BSE in a mouse system, it is possible that there is an as yet symptom- free cohort of infected individuals that are still incubating vCJD. Although it is considered highly unlikely, the possibility of human-to-human transmission remains.