Central sensitization
In the spinal cord central sensitization arises from changes in the behavior of dorsal horn cells (DHCs) in response to prolonged nociceptor traffic. This strengthens (facilitates) the synapses onto the DHCs. The release of substance P from C fiber nociceptor terminals generates slow epsps. If C fibers are stimulated at a constant frequency above 0.3 Hz these epsps summate to produce ever-larger depolarization of the DHCs, which activates NMDA receptors. This is a type of temporal summation called wind-up which is as shown in the figure below. This activity-dependent synaptic facilitation takes only seconds to induce but NMDA receptor activation switches on a cascade of kinase activation that produce persistent increases in excitability by, for example, recruiting more AMPA glutamate receptors, and the formation of new synapses. Central sensitization has many similarities to long term potentiation, the synaptic plasticity that underlies memory.
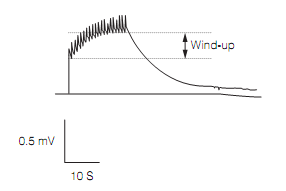
Figure: Wind-up in extracellular recording of spinal cord local field potential in response to C-fiber stimulation at 1 Hz.
The wide dynamic ranges DHCs also become hyperexcitable to low threshold (Aβ fiber) input and increase their receptive field size. This underlies tactile allodynia in which touch is painful. Under normal circumstances stimuli applied near the edge of the receptive field of a DHC usually elicit subthreshold responses (epsps). However, if a cell is much depolarized it is more sensitive to excitatory input. Previously subthreshold stimuli at the edge of its receptive field now become suprathreshold; in effect the receptive field is enlarged.Analgesic cover in surgery is optimal if given preemptively and topped-up frequently enough that no pain occurs. This prevents wind-up and central sensitization.
Central sensitization is not confined to the spinal cord. It also occurs in the rostroventral medulla, thalamus, amygdala, and cingulate cortex.
Central sensitization in chronic inflammatory disorders involves glial cells as shown in figure below. Peripheral nerve injury activates microglia in the local spinal cord. This up-regulates microglial P2X4 receptors, one of a family of ligand-gated ion channels which respond to ATP. Stimulation of the P2X4receptors by the excessive ATP released by damaged primary afferents (ATP is a fast transmitter in many afferents) causes the microglia to release cyto kines which increase the sensitivity of lamina I dorsal horn cells. This happens becausethe cytokines down-regulate an anion exporter in the lamina I cell bringing about a large rise in intracellular Cl- concentration. These cells now respond to activation of their GABAA and glycine receptors by a large depolarization, caused by Cl- leaving the cell down its concentration gradient. The depolarization permits NMDA receptors to be opened by any incoming excitation, triggering potentiation.

Figure: A model for central sensitization in response to peripheral nerve injury. Numbers indicate the series of events. The Glutamatergic input from remaining intact fibers because prolonged firing of the lamina I cell because the voltage-dependent blockade on its NMDA receptors has been removed by the abnormal depolarization (8).
Microglias are also involved in central sensitization in the thalamus. Here a cytokine released by thalamic neurons activates COX2 in the glia so they synthesize and secrete prostaglandin E2. This binds to prostaglandin receptors on neurons and increases their excitability by depolarization.