Hemoglobino-pathies
Comparison of primary sequences of hemoglobin chains from over 60 different species reveals that only 9 residues in polypeptide chain are
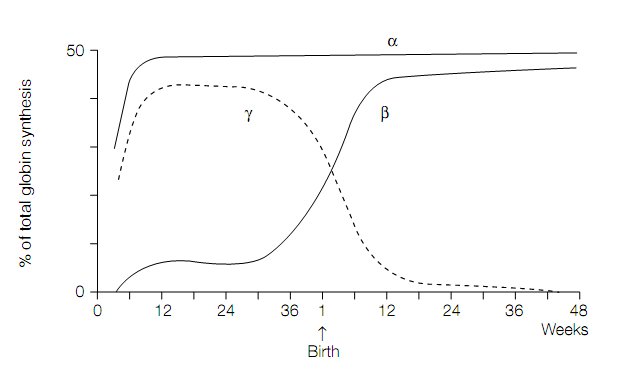
Figure. The switch in human globin chain synthesis at the time of birth.
invariant (that is the same) between all the species. These 9 residues include proximal and distal histidines that are essential for the correct functioning of the protein. Many of the other residues can be replaced from one species to the other by residues with the similar properties (for example the hydrophobic valine is replaced with hydrophobic isoleucine, or polar serine is replaced with polar asparagine), so-called conservative substitutions. On the contrary only a few residues have changed between the species to a completely different residue (such as a hydrophobic leucine to positively charged lysine or negatively charged glutamate to positively charged arginine), called as nonconservative substitutions, as this type of change could have a major effect on structure and function of protein.
Several hundred abnormal hemoglobins have been characterized, which gives rise to the so-called hemoglobinopathies. Probably the best characterized hemoglobin-pathy is sickle-cell anemia (sickle-cell hemoglobin; HbS). This disease can be characterized by the patient’s erythrocytes which has characteristic sickle or is having cres cent shape. The basis for this disease is change of a glutamic acid residue for valine at position 6 of β-chain, resulting in substitution of a polar residue by the hydrophobic one. The nonconservative substitution of the valine for glutamate provides HbS a sticky hydrophobic patch on outside of each of its β-chains. In corner between helices E and F of β-chain of deoxy-HbS is hydrophobic site which is complementary to sticky patch.
Thus the complementary site on one deoxy-HbS molecule is bind to the sticky patch on the other deoxy-HbS molecule, resulting in formation of long ?bers of hemoglobin molecules which distort the erythrocyte. Electron microscopy has revealed that ?bers have diameter of 21.5 nm and consist of 14-stranded helix. Multiple polar interactions, in addition to critical interaction between sticky patches, stabilize the ?ber. In oxy-HbS complementary site is masked, so the formation of long ?bers occurs when there is high concentration of deoxygenated form of HbS.
Sickle-cell anemia is genetically transmitted, hemolytic disease. The sickled cells are much fragile than normal erythrocytes, lysing much easily and having a shorter half-life, which leads to severe anemia. As sickle-cell anemia is transmitted genetically, homozygotes have two copies of abnormal gene while heterozygotes have one abnormal and normal copy. Homozygotes
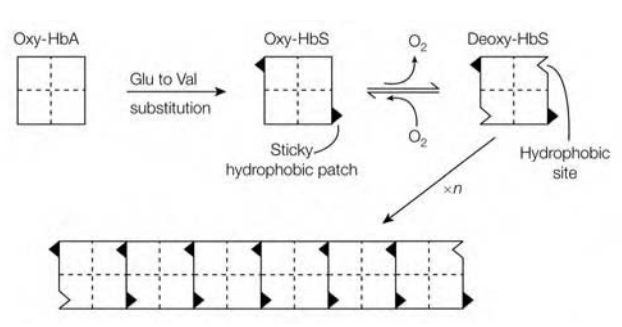
Figure:Molecular basis for the aggregation of deoxyhemoglobin molecules in sickle-cell anemia.
have a reduced life-span often as a result of renal failure, infection, cardiac failure or thrombosis, because of the sickled cells becoming trapped in small blood vessels leading to tissue damage. On the contrary, heterozygotes are not symptomatic usually as only approximately 1 percent of their erythrocytes are sickled, compared with approximately 50 percent in a homozygote. The frequency of the sickle gene is high in certain parts of Africa and correlates with incidence of malaria. The reason for this is that the heterozygotes are protected against most lethal form of malaria, while normal homozygotes are vulnerable to the disease. Inheritance of abnormal hemoglobin gene can now be monitored by recombinant DNA methods.