Ionotropic glutamate receptor family
The ionotropic glutamate receptors (iGluRs) have a tetrameric quaternary structure and only share weak homologies with the cys-loop family. There are three populations of iGluRs, defined by the selective agonists; the AMPA receptors (named after α-amino-3-hydroxy-5methyl-4-isoxazole proprionic acid), the kainate receptors and NMDA receptors (NMDARs). All are fairly nonselective cation channels; however NMDA receptors and a few AMPA receptors favor calcium permeation.
The secondary structure of the AMPA and kainate receptor subunits which is as shown in figure below has four membrane-spanning segments (M), one of which (MII) is on a re-entrant loop that contributes to the pore. The MII-consisting part of the molecule has a striking resemblance to the S5-H5-S6 area of potassium channels, albeit oriented in the membrane in the reverse sense. These receptors may therefore have evolved from ancient potassium channel.
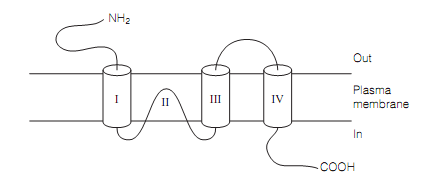
Figure: Ionotopic glutamate receptor subunit structure.
The four subunits, GluR1–GluR4, contribute to the AMPA receptors. Nearly all are assembled from GluR2 subunits altogether with either GluR1 or GluR3 subunits. The GluR2 subunits have numerous instead of special roles in the AMPA receptors, it do the following:
- Manage the voltage gating of the receptor
- Come in two forms which differ in their MII area by merely one amino acid (this modifiesthe receptor from Na+ and K+ channel to the Ca2+ channel)
- Control the transport of the AMPA receptors to postsynaptic membrane
- Encourage the growth of dendritic spines on the cortical pyramidal cells.
The NMDA receptor is named after the selective agonist N-methyl-d-aspartate. Its natural agonists are aspartate and glutamate. The receptor is significant as it is implicated in key aspects of brain function like learning, development, and memory, and in pathologies, for illustration strokes and epilepsy.
The NMDA receptors are heterotetramers consisting of two NR1 and two NR2 subunits. There are eight NR1 isoforms formed by alternative splicing of a single gene and four NR2 isoforms. The NR1 subunits are expressed ubiquitously in the brain while NR2 isoforms are expressed in a area-specific way. This gives rise to multiple NMDA receptors with different brain distributions and functional properties occuring from the specific combination of the subunits. There are also two NR3 isoforms which confer inhibitory properties on NMDA receptors. The secondary structure resembles the figure shown below. The extracellular N-terminal domain of the NR2 subunits binds the neurotransmitter whereas the extracellular domain between MIII and MIV is a modulatory domain which binds the co-agonists glycine or serine. There are structural similarities among the agonist-binding domain of the NR2 subunits and those of the other ionotropic glutamate receptors and with ancient amino acid-binding proteins of bacteria. The extensive cytoplasmic domain has phosphorylation sites and areas for binding the structural proteins.
The NMDA receptors have few unusual properties which are summarized below:
- The Glycine and D-serine act as allosteric co-agonists to potentiate the effects of the glutamate. D-serine is synthesized and released by the astrocytes co-localized with neurons consisting of NMDA receptors, might also be released by the neurons, and might be the primary ligand at the co-agonist site.
- The NMDA receptors are voltage-gated. The resting membrane potentials glutamate will bind to the receptor however the ion channel is blocked by Mg2+ ions. This blockade is lifted only by a large depolarization. In another words, the ion channel is opened only if glutamate binds and the receptor experiences the depolarization at similar time. This behavior is critical to NMDA receptor functions.
- The ion channel is permeable to Ca2+ and also Na+ and K+. The activation of NMDA receptors raises intracellular Ca2+ concentrations and this can activate numerous second messenger cascades, many of which modify AMPA receptor functions. This is vital for long-term potentiation (LTP) and long-term depression (LTD).
- Most of the NMDA receptor kinds are inhibited by H+ and represent partial inhibition at physiological pH.
- They hold a redox modulatory site where reductants enhance and oxidants depress NMDA channel activity. This site is presumably the aim for endogenous redox agents like glutathione.
- The channel is blocked by Zn2+ ions, and Pb2+ is a potent antagonist.
- The NMDA receptors are aim for dissociative anesthetics (example, ketamine), nitrous oxide, many opiates, and for the psychotomimetic drug phencyclidine.