MOLECULAR ORBITALS: HOMONUCLEAR DIATOMICS
In chemistry, a molecular orbital (or MO) is a mathematical function explaining the wave-like behaviour of an electron in a molecule. This function can be employed to calculate physical and chemical properties like the probability of finding an electron in any particular region. The word "orbital" was first time used in English by the Robert S. Mulliken as the English translation of Schrödinger's 'Eigenfunktion'. After that it has since been equated with the "region" generated with the function. Molecular orbitals are generally constructed by combining atomic orbitals or hybrid orbitals from every atom of the molecule or other molecular orbitals from groups of atoms. By using the Hartree-Fock or Self-Consistent Field (SCF) methods they can be quantitatively calculated.
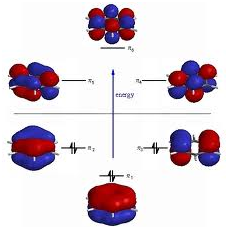
Molecular orbitals (MOs) depict areas in a molecule where an electron is expected to be found. Molecular orbitals are achieved from the combination of atomic orbitals that predict the location of an electron in an atom. A molecular orbital can state that the electron configuration of a molecule: the energy and spatial distribution of one (or one pair of) electron(s). Generally an MO is represented as a linear combination of atomic orbitals (the LCAO-MO method), particularly in qualitative or very approximate use. They are invaluable in providing an easy model of bonding in molecules, understood from molecular orbital theory. Most present-day techniques in computational chemistry begin by calculating the MOs of the system. A molecular orbital explains the behavior of one electron in the electric field generated by the nuclei and some average distribution of the other electrons. In the case of two electrons occupying similar orbital, the Pauli principle demands that they have reverse spin. Essentially this is an approximation and highly accurate explainations of the molecular electronic wave function do not have orbitals.