There are many characteristics of the ideal drug candi-date. One of these is that its pharmacokinetics should meet its intended use. Many potentially useful drug can-didates, however, fail because the molecule has undesir-able pharmacokinetic properties, such as poor bioavail-ability, thereby limiting oral administration, or poor metabolic stability, thereby severely limiting the possi-bility of once-daily administration. In the following paragraphs, a proposed strategy for model building in the very early phase of drug candidate selection is ex-plored. This strategy is based on the separate simulation of the quantitative features of absorption, distribution, and elimination processes, followed by the integration of these 3 elements into a single PBPK model.
Early Candidate Selection
Ideally, it would be beneficial to be able to predict the pharmacokinetic behavior of a compound ab initio from its "molecular descriptors" (eg, lipophilicity, molecular size and shape, charge density distribution) alone, as then only compounds with desirable features would be synthesized and investigated further. Although not cur-rently, if ever totally, realizable, nonetheless such in silico-generated descriptors may enable a reasonable initial prediction of the likely behavior in vivo of some processes, in particular those based on passive processes such as diffusion and simple partitioning (eg, into fat). However, the task of predicting the likely overall phar-macokinetic behavior in vivo is becoming more feasible by coupling in silico computations with pertinent in vitro data, such as plasma protein binding, microsomal or hepatocyte intrinsic clearance, and cell membrane per-meability (eg, across Caco2 cells), which allow for the inclusion of active processes involved in metabolism and membrane transport. Using mechanistically based software (see Appendix 2) estimates can be made as to the expected rate and extent of absorption, the affinity of compound for individual tissues, and hepatic clearance, which when placed appropriately within a "generic whole body PBPK model" allows prediction of the tem-poral profile of a compound in both plasma and tissues. Verification and further refinement of this modeling ap-proach can only be performed in animals, although often the ultimate objective is to predict likely profiles in hu-mans.
As the required input parameters can be generated dur-ing early discovery, the application of generic PBPK models becomes feasible during the clinical candidate optimization and selection process. Such implementa-tion would have several advantages: (1) it would aid in the candidate selection itself, as mentioned above, by improving the likelihood of selecting compounds with desirable PK properties; (2) it would result in a reduction in unnecessary animal testing, as it may well avoid the testing of compounds whose PK properties in humans are predicted to be inadequate for intended use; (3) being mechanistically based, it would contribute to a system-atic and rational approach for identification of the key parameters of a compound that should be defined in early development, as it can be very expensive to first become aware of these much later in drug development; (4) the overall PK of potential drug candidates in ani-mals (and humans, made possible through a combination of scaling of the physiological parameters and use of in vitro human data within the frame of the whole body PBPK construct) can be anticipated prior to any in vivo experiments, thereby helping to improve the design of such studies; and (5) it would improve the ability to more reliably extrapolate PK across species (see Figure 2), routes of administration, and dose levels. During the development process, additional data (eg, tissue kinetics during safety assessment) may be generated that can be used to improve the generic PBPK models by adding information and/or replacing in silico and in vitro input data. In this sense the in silico and in vitro-based PBPK modeling approach is complementary to the conven-tional whole body PBPK approach, which requires in vivo tissue kinetic data as input information.
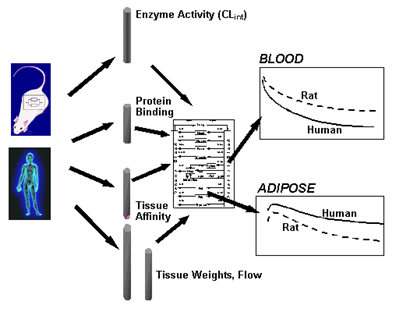
Figure Schematic illustrating how differences in the magnitude of various parameters, such as intrinsic meta-bolic activity, plasma protein binding, tissue affinities, tissue volumes, and blood flow, propagated through a whole body physiologically based model, explain the differences in the concentration-time profiles in blood and tissues between rat (or any other animal) and hu-man.
Preclinical Phase
There is a major push to predict the likely pharmacoki-netic performances of drugs in humans from preclinical data, thereby helping to avoid evaluation in humans of compounds with poor pharmacokinetic performance, which can severely limit their practical use. Here, scal-ing forms an important element of the process. The al-lometric approach, which assumes that any differences across species are driven by body size alone, has long been the dominant method for interspecies scaling. However, due to the failure to account for differences in active processes, such as metabolism and transport, the use of this approach to predict the "first into human strategy" has come under significant criticism. At pre-sent, the decision is often taken based on toxicological and animal pharmacological considerations alone. From the available experience, it clearly appears that PBPK modeling offers a modern, science-founded approach to help rationalize the "first into human" decision-making process. By challenging the observed human data against predictions over many compounds, increasingly more accurate predictive PBPK models will be pro-duced, although this approach is likely to be most suc-cessful within a series of structurally related compounds, as commonly arises in drug development. PBPK model-ing also has the potential to explain certain aspects of species differences in nonclinical safety studies, such as accommodating for differences in tissue-specific trans-porters and binding constituents as well as in relative composition of body fat.
As clinical and commercial interest in a particular com-pound increases so does the range of studies surrounding it, including in vivo animal tissue distribution studies, as part of safety assessment. Here the opportunity arises to evaluate the ability of in silico and in vitro methods to predict tissue distribution, as well as to explore such facets as concentration dependence and permeability. Studies to date show that in many circumstances the distribution of a compound into a particular tissue is similar in animals and humans, which would suggest that the components of the tissue that are responsible for its distribution are common across the species, including their relative composition. Indeed, this is an almost universal assumption in the scaling of animal data to predict pharmacokinetics in humans, whether working with drug substances or environmental compounds. This assumption may not always hold, as often reflected by poor prediction of the volume of distribution of the compound in humans, even after correcting for differences in plasma protein binding across species. When the assumption fails, it is important that the reason for it be pursued, as the failure may be due to species differences in tissue composition of the primary binding constituents, which once characterized would allow cross-species prediction of tissue distribution for related com-pounds. Also, in vitro studies with human tissues, which have increasingly become available (liver being a well-known but by no means the only example), would allow greater study of the relevance of animal data to predict tissue distribution in humans, currently a relatively neglected area compared with in vitro studies of metabolism and absorption.